

漢方薬滅菌器
Cat:製品
飽和蒸気を滅菌媒体として湿熱滅菌を行う滅菌器で、脈動真空機能、冷気干渉除去機能、真空乾燥機能を備えています。 漢方工学の分野において、中医学(中医学)の粉末、煎じ顆粒、錠剤、粉末などの...
詳細を見る
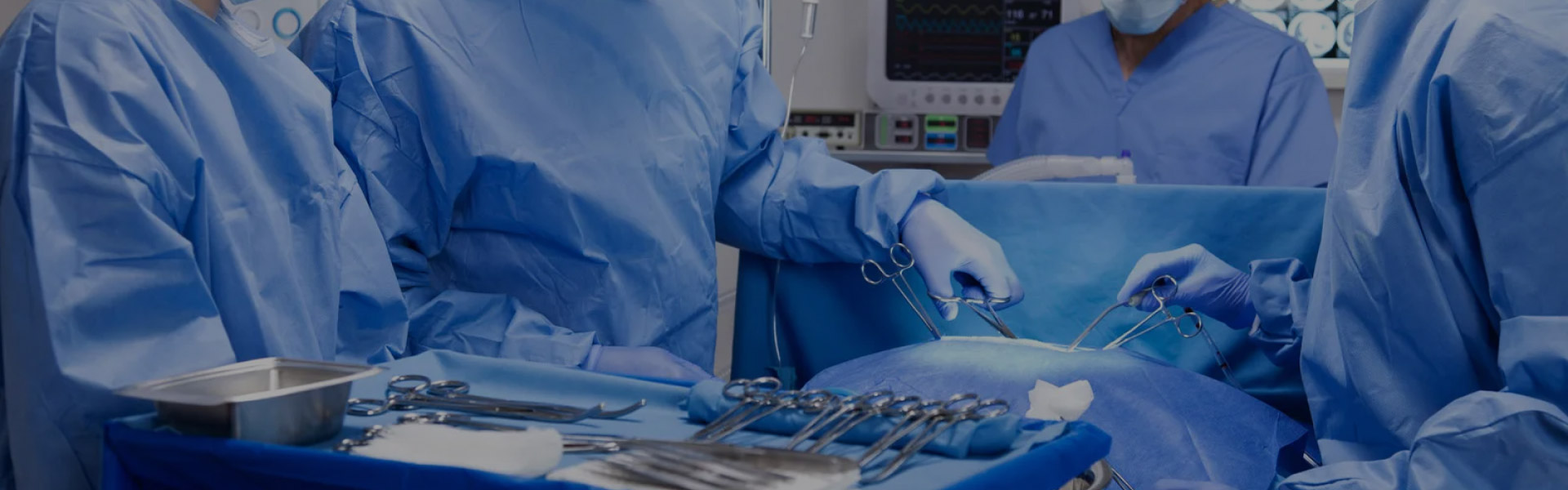
無菌調製とは、生存微生物を含まない製品である無菌製品を配合または製造するプロセスを指します。これは、注射剤、点滴静注液、点眼液など、人体への投与を目的とした製品を製造する業界では重要なプロセスです。また、手術やその他の処置で使用される滅菌医療機器や器具を準備するためにも不可欠です。
製薬、医療、研究施設における滅菌の重要性
滅菌は、細菌、ウイルス、真菌、胞子など、あらゆる形態の微生物を除去または不活化するプロセスです。製薬、医療、研究室の現場では、交渉の余地はありません。適切な滅菌を行わないと、製品や機器に有害な微生物が潜伏し、患者の感染、製品の汚染、不正確な検査結果を引き起こす可能性があります。これは患者の安全を危険にさらすだけでなく、企業の財務上および評判上の重大な損害につながる可能性があります。
無菌製品の安全性と有効性を確保するために、厳格な規制と基準が設けられています。の FDA の適正製造基準 (GMP) ガイドラインは、無菌医薬品を製造するための枠組みを提供します。の 米国薬局方 (USP) などの特定の章の概要を説明します。 USP<797> 配合された無菌製剤については、無菌性保証の要件が詳しく説明されています。さらに、 ISO規格 、みたいな ISO11135 エチレンオキシド滅菌用および ISO11137 放射線滅菌については、滅菌プロセスに関する国際的に認められたガイドラインを提供します。これらの規制と基準は、使用する機器から滅菌プロセスの検証と文書化に至るまで、あらゆるものを規定します。
処理される材料の性質、熱や湿気に対する感受性、必要な滅菌保証レベルに基づいて、さまざまな滅菌方法が選択されます。
オートクレーブは、最も一般的で信頼性の高い滅菌方法です。 湿った熱 の形で 加圧飽和蒸気 .
蒸気滅菌のしくみ
このプロセスは、加圧下でアイテムを高温の蒸気にさらすことによって機能します。乾燥空気よりも熱伝達率が高い蒸気からの熱は、微生物内のタンパク質の不可逆的な凝固と変性を引き起こし、微生物を効果的に死滅させます。圧力により蒸気が通常の沸点を超える温度に達し、滅菌時間が大幅に短縮されます。一般的なサイクルには、チャンバーから空気を除去する調整段階、品物を指定された期間目標の温度と圧力に保持する暴露段階、および圧力を解放して負荷を乾燥させる最終排気段階が含まれます。
アプリケーションとユースケース
蒸気滅菌は以下の用途に最適な方法です。 熱や湿気に強いアイテム 、次のような:
手術器具
ガラス製品
カルチャーメディア
テキスタイルとリネン
水溶液および液体
乾熱滅菌用途 熱風 ~のプロセスを通じて微生物を殺す 酸化 .
乾熱滅菌の仕組み
凝固に依存する湿熱滅菌とは異なり、乾熱滅菌は微生物を死滅させます。 細胞成分の酸化 そして タンパク質の変性 。この方法は蒸気よりも熱伝達効率が低く、同じレベルの無菌性を達成するにはより高い温度とより長い曝露時間を必要とします。
アプリケーションとユースケース (e.g., Depyrogenation)
この方法は、蒸気に耐えられない湿気に敏感で熱に安定な素材や、発熱物質(発熱を引き起こす物質、多くの場合細菌性エンドトキシン)の除去に適しています。乾熱滅菌に必要な高温は、「乾熱滅菌」として知られるこのプロセスに特に効果的です。 脱発熱物質 .
一般的なアプリケーションには次のものがあります。
ガラス製品 (vials, ampoules)
油脂、粉末
湿気の存在下で腐食する特定の金属器具
気相滅菌は、 低温法 ガス状の化学薬品を使用して微生物を不活化するものです。
気相滅菌の概要
このプロセスには、次のような液体滅菌剤の変換が含まれます。 過酸化水素 、チャンバーを満たす蒸気になります。蒸気は負荷に浸透し、必須の細胞成分の酸化を通じて微生物を破壊します。多くの場合、プラズマ相と組み合わせて蒸気を水や酸素などの無毒な副産物に分解し、エアレーションの必要性を排除します。
アプリケーションとユースケース
気相滅菌は次の目的で使用されます。 熱や湿気に弱い医療機器 オートクレーブ滅菌すると損傷する可能性があります。その低温サイクルは以下の用途に最適です。
複雑な内腔を備えた医療機器
電子機器およびその他の機密機器
使い捨て医療機器
ろ過 is a 非加熱滅菌法 液体や気体に使用されます。
無菌処理におけるフィルターの使用
この方法では、細菌やその他の微生物を保持するのに十分な細孔サイズのフィルターに液体を通過させることにより、液体から微生物を物理的に除去します。これは、制御された環境で無菌コンポーネントから無菌製品を製造する無菌処理における重要なステップです。濾過では微生物は死滅しません。単にそれらを削除するだけです。
フィルターの種類と選択
滅菌グレードのフィルターの孔径は通常、 0.22ミクロン以下 バクテリアの保持を確実にするため。フィルタの選択は、流体の特性とプロセス要件によって異なります。
メンブレンフィルター 滅菌に使用される最も一般的なタイプは、ポリエーテルスルホン (PES) やポリフッ化ビニリデン (PVDF) などのポリマーで作られています。
深度フィルター 最終的な滅菌グレードのメンブレンフィルターの前に、大きな粒子を除去するためのプレフィルターとして使用されることがあります。
| 滅菌方法 | 作用機序 | 代表的な温度範囲 | 標準的なサイクルタイム | 主な用途 |
| 蒸気滅菌 | 湿熱によるタンパク質の変性 | 121°C ~ 134°C (250°F ~ 273°F) | 15~30分(121℃)または3~10分(134℃) | 熱や湿気に強いもの、多孔質材料、水溶液 |
| 乾熱滅菌 | 熱風による細胞成分の酸化 | 160°C ~ 250°C (320°F ~ 482°F) | 1~2時間以上(170℃) | 熱に安定で湿気に敏感なアイテム、ガラス製品の発熱物質除去 |
| 気相滅菌 | 化学蒸気による酸化 | 37°C ~ 60°C (98°F ~ 140°F) | 30~90分(サイクルにより異なります) | 熱や湿気に弱い医療機器、電子機器 |
| ろ過 | 微生物の物理的除去 | 流体の周囲温度から使用温度まで | 流量と体積によって異なります | 熱に弱い液体および気体 (医薬品、滅菌空気など) |
適切な滅菌器の選択は、安全性、コンプライアンス、運用効率に影響を与える重要な決定です。それは画一的な選択ではなく、さまざまな要因によって決まります。
アイテムの材質の適合性が最も重要な要素です。
金属器具、ガラス製品、水溶液などの熱や湿気に安定したアイテムは、蒸気滅菌に最適です。
油、粉末、特定のガラス製品など、熱に安定ではあるが湿気に弱いものは乾熱滅菌が必要です。
複雑な電子機器やプラスチックを備えた多くの最新の医療機器など、熱や湿気に弱い素材は、気相滅菌などの低温方法に最適です。
間違った方法を使用すると、製品が損傷したり、滅菌できなくなったり、あるいはその両方になる可能性があります。
各滅菌方法には、必要な滅菌保証レベル (SAL) を達成するために満たさなければならない特定のパラメーターのセットがあります。
蒸気滅菌の場合、重要なパラメータは温度、圧力、時間です。これらは相互に依存しています。温度が高いほど、暴露時間は短くなります。
乾熱滅菌では、熱風の熱伝達効率が低いため、はるかに高い温度と長時間が必要になります。
気相滅菌は、温度、滅菌剤(過酸化水素など)の濃度、時間の正確なバランスに依存します。
これらのパラメータは細心の注意を払って検証されており、すべてのサイクルで正確に従う必要があります。
滅菌が必要なアイテムの量と必要な処理速度によって、必要な滅菌器のサイズとタイプが決まります。
大規模な医薬品製造には、大容量の自動化システムが不可欠です。
小規模な研究室やクリニックでは、コンパクトな卓上オートクレーブのみが必要な場合があります。
サイクル タイムはスループットの重要な要素です。たとえば、蒸気滅菌は一般に乾熱滅菌よりも高速ですが、特定の品目については蒸気相滅菌の方が非常に迅速な対応が可能です。
規制産業におけるすべての滅菌プロセスは、必要な SAL を一貫して達成していることを証明するために検証される必要があります。
これには、GMP、USP、ISO 規格を満たすための一連のテストと文書化が含まれます。
滅菌器の設計、制御、および監視機能は、これらの検証活動をサポートする必要があります。
検証の複雑さとコストは、滅菌方法が異なると大きく異なる場合があります。
総所有コストは機器の初期購入価格を超えます。
初期コスト: 乾熱滅菌器や一部の小型オートクレーブは安価ですが、大規模な気相システムや特殊なオートクレーブには多額の投資が必要です。
運営コスト: これには、光熱費(電気、水、蒸気)、消耗品(滅菌剤、梱包、生物学的インジケーター)、メンテナンスが含まれます。蒸気滅菌は通常、気相法よりも消耗品のコストが低くなります。
人件費や失敗したサイクルのコストも考慮する必要があります。
コストとパラメータの比較
| 考察 | 蒸気滅菌 (Autoclave) | 乾熱滅菌 | 気相滅菌 |
| 初期費用 | 中程度から高程度 | 低から中程度 | 高 |
| 運用コスト | 少ない(水道、電気) | 低い(電力) | 中程度から高程度 (Consumables, specialized sterilant) |
| 典型的な温度 | 121℃~134℃ | 160℃~250℃ | 37℃~60℃ |
| 標準的なサイクルタイム | 15~60分 | 1~4時間 | 30~90分 |
| 材質の適合性 | 熱と湿気に安定 | 熱に安定、湿気に弱い | 熱と湿気に敏感 |
滅菌プロセスを成功させるには、単にアイテムを滅菌器に入れるだけでは不十分です。一貫した無菌結果を保証するために、細心の注意を払った準備、適切な充填、継続的なモニタリング、および慎重なサイクル管理が必要となります。
効果的な滅菌は徹底的な準備から始まります。アイテムは滅菌する前に洗浄し、除染する必要があります。血液や組織などの残留有機物はバリアとして機能し、微生物を滅菌剤から守るため、サイクルの失敗につながります。
クリーニング: これは最初で最も重要なステップです。これには、洗剤、水、および機械的作用を使用して、目に見えるゴミを物理的に除去することが含まれます。超音波洗浄機は、複雑な部品を備えた器具によく使用されます。
乾燥: 洗浄後は、アイテムを完全に乾燥させる必要があります。湿気は乾熱などの滅菌プロセスを妨げる可能性があり、蒸気滅菌の場合、過剰な水は非滅菌とみなされ湿ったパックを引き起こす可能性があります。
包装:商品は、サイクル後に無菌性を維持しながら、滅菌剤(蒸気、蒸気など)の浸透を可能にする材料で包装されます。一般的なパッケージには、滅菌パウチ、ラップ、硬質容器が含まれます。パッケージは密封され、正しくラベルが貼られている必要があります。
滅菌器のロードとアンロード
滅菌剤がすべての品目のすべての表面に確実に届くようにするには、適切な充填が不可欠です。装填が正しくないと、滅菌が達成されない「コールド スポット」が発生する可能性があります。
積載: アイテムはチャンバー内にゆったりと配置し、空気の除去と蒸気または蒸気の循環のための十分なスペースを確保する必要があります。滅菌器に過負荷をかけないでください。重いものは下の棚に置く必要があります。蒸気滅菌器の場合、水が溜まる可能性のあるアイテム (ボウルなど) は、排水できるように傾ける必要があります。
取り外し: サイクルが完了したら、滅菌器が冷えて圧力が均等になるまで待ってからドアを開けてください。荷降ろしの際は、パックを慎重に取り扱い、濡れや損傷の兆候がないか検査してください。ウェットパックは再処理する必要があります。
監視および制御システムは、滅菌プロセスが正しく進行していることをリアルタイムで保証します。
物理的モニタリング: これには、滅菌器の内部ゲージと表示値をチェックして、プログラムされた温度、圧力、時間のパラメーターが満たされていることを確認することが含まれます。
化学インジケーター: これらは、特定の滅菌パラメータにさらされると色や形状が変化する材料です。これらにより、アイテムが滅菌剤にさらされたことを視覚的に迅速に確認できます。化学インジケーターには、暴露を示す単純なテープから、複数の重要なパラメーターを検証する統合インジケーターまで、さまざまな種類があります。
生物学的指標 (BI): 最も確実なモニタリング方法。 BI には耐性の高い細菌胞子が含まれています。滅菌プロセスが成功すると、胞子は死滅します。次に、BI を培養します。微生物の増殖がなければ、滅菌サイクルが成功していることが確認されます。
サイクル開発は、新しい製品や新しい滅菌器など、新しい滅菌プロセスの特定のパラメーターを定義するプロセスです。これは検証プロセスの重要な部分です。
目標は、必要な無菌保証レベル(SAL)を一貫して提供する、堅牢で反復可能なサイクルを確立することです。
最適化は、たとえば、無菌性を維持しながらサイクル時間を短縮することにより、検証されたサイクルの効率を向上させることを目的としています。これには、生物学的および化学的指標を使用した広範なテストに基づいたパラメータの微調整が含まれることがよくあります。
検証は、滅菌プロセスが、所定の仕様および品質特性を満たす製品を一貫して生産することを確認する文書化されたプロセスです。規制された業界では、患者の安全と製品の有効性を確保するための基本的な要件です。
滅菌は「特別なプロセス」であり、最終製品を検査することによってその結果を完全に検証することはできません。無菌に見える製品は、破壊試験を行わなければ無菌であることを証明できません。したがって、無菌性に対する信頼は、検証され管理されたプロセスを通じて得られます。検証により、滅菌器とその関連プロセス パラメーターが 10-6 以上の滅菌保証レベル (SAL) を一貫して提供できるという科学的証拠が得られます。これは、単一の非滅菌ユニットが発生する確率が 100 万分の 1 未満であることを意味します。
滅菌検証は通常、次の 3 つの部分からなるプロセスです。
設置適格性評価 (IQ): 滅菌器がメーカーの仕様に従って正しく設置されていることを検証します。これには、機器が適切にセットアップされ、安全に動作することを確認するための、文書、ユーティリティ (電力、水道、蒸気)、および物理的な設置のチェックが含まれます。
動作適格性評価 (OQ): このステップでは、滅菌器の動作限界と、指定されたパラメータ内で機能する能力をテストします。 OQ は、最高温度と最低温度、最長と最短のサイクル タイムなどの「最悪の場合」のシナリオで機器をテストし、それでも望ましい結果を達成できることを確認します。これにより、滅菌器の安全かつ効果的な動作範囲が確立されます。
パフォーマンス適格性評価 (PQ): 最後の最も重要なステップ。 PQ は、特定の製品とそのパッケージを含む滅菌プロセス全体で一貫して滅菌出力が生成されることを実証します。これは、訓練を受けた担当者を使用して、全負荷をかけた通常の動作条件下で実行されます。このフェーズでは、プロセスが堅牢であり、実際の運用環境で再現可能であることが証明されます。
検証と日常的な監視は、これらの主要なツールに依存して、サイクルが成功していることの証拠を提供します。
生物学的インジケーター(BI): これらは滅菌を証明するためのゴールドスタンダードです。 BI には、担体材料 (紙片など) 上に高耐性細菌胞子の既知の集団が含まれています。使用される胞子の種類は滅菌方法によって異なります。たとえば、Geobacillus stearothermophilus は蒸気と過酸化水素蒸気に使用され、Bacillus atrophaeus は乾熱とエチレンオキシドに使用されます。サイクル後に胞子が死滅した場合、そのプロセスがすべての微生物にとって致死的であったという高い確信度が得られます。
化学インジケーター(CI): これらは、特定のパラメーターが満たされたことを示す視覚的なフィードバックを即座に提供します。
クラス 1 (プロセスインジケータ): アイテムが滅菌プロセスにさらされたことを単に示すオートクレーブテープ。
クラス 4 (マルチパラメーター インジケーター): 温度や時間などの 2 つ以上の重要な変数に反応するストリップ。
クラス 5 (統合インジケーター): これらは生物学的インジケーターのパフォーマンスと相関し、サイクルのすべての重要なパラメーターに反応するように設計されています。
IQ から PQ に至る検証プロセスの各ステップは、徹底的に文書化する必要があります。これには、プロトコル、テスト結果、逸脱、最終検証レポートが含まれます。細心の注意を払って記録を保管することは規制要件 (GMP、ISO) であり、監査中にコンプライアンスを証明するために不可欠です。記録は滅菌器とそのパフォーマンスの完全な履歴を提供し、トレーサビリティと説明責任を保証します。
滅菌器の長期的な信頼性と安全性を確保するには、定期的なメンテナンスが不可欠です。プロアクティブなケアは、一貫したパフォーマンスを保証するだけでなく、費用のかかる故障を防ぎ、機器の寿命を延ばすのにも役立ちます。
定期的なメンテナンス作業
明確に定義されたメンテナンス スケジュールが重要です。タスクは、日次、週次、月次のルーチンに分類できます。
毎日: 残留物の蓄積を防ぐために、チャンバー、ドアのガスケット、外面を掃除してください。目に見える損傷や漏れがないか確認してください。
毎週: ドアのシールと排水フィルターを点検して掃除します。チャンバーを徹底的に掃除して、蒸気の浸透を妨げる可能性のあるスケールや破片を取り除きます。
毎月: タンクの清掃や安全機能と制御装置が適切に機能しているかどうかの確認など、より詳細な清掃を実施します。
校正は、滅菌器の機器(温度センサーや圧力センサーなど)を調整して、正確な測定値が得られるようにするプロセスです。これは、資格のある技術者が、校正された追跡可能な機器を使用して、定期的、計画的に、多くの場合は年に一度実行する必要があります。
保守には、滅菌器の機械システムと電気システムの専門的かつ包括的な検査が含まれます。これには、ドアの機構、ポンプ、バルブ、発熱体のチェックが含まれます。通常、年に 1 ~ 2 回、専門家による定期的なサービスを受けると、重大な障害につながる前に軽微な問題を特定して修正できます。
| 問題 | 潜在的な原因 | 解決策 |
| ウェットパック | チャンバーに過負荷がかかる。不適切な梱包。ドレンフィルターの詰まり。乾燥サイクルに失敗しました。 | チャンバーに詰め物を詰めすぎないでください。適切な滅菌包装を使用する。排水フィルターを掃除します。乾燥サイクルパラメータを確認してください。 |
| 滅菌失敗 | サイクルパラメータが正しくありません。滅菌器の故障(温度センサーの故障など)。チャンバー内のエアポケット。 | サイクル設定とロード手順を確認します。 Bowie-Dick テストを実行します (真空前蒸気滅菌器の場合)。専門家にユニットの修理を依頼してください。 |
| ドアガスケットの漏れ | ガスケットが損傷または汚れている。ガスケットが正しく取り付けられていません。過度の摩耗。 | ガスケットとシール面を清掃します。ガスケットが損傷または摩耗している場合は交換してください。 |
| スケールまたは残留物の蓄積 | 非蒸留水または硬水を使用しています。 | 蒸留水または脱イオン水のみを使用してください。メーカーの推奨に従って、スケール除去剤を使用して定期的なチャンバーのクリーニングを実行してください。 |
適切なケアとメンテナンスにより、滅菌器の寿命を大幅に延ばし、投資を保護し、長期的な信頼性を確保できます。
正しい水質を使用してください。蒸気滅菌器の場合は、パイプの詰まりや発熱体に損傷を与える可能性のある鉱物の堆積を防ぐために、蒸留水または脱イオン水のみを使用してください。
メーカーのガイドラインに従う: 滅菌器のマニュアルに指定されている、洗浄、メンテナンス、および操作に関するすべての指示に従ってください。
予防メンテナンスを実行します。問題が発生するまで待つ必要はありません。プロアクティブなメンテナンス スケジュールは、事後対応の修理よりも効果的で、コストが低くなります。
担当者を徹底的に訓練する: すべてのオペレーターが正しい積載手順、日常のケア、エラーや故障が発生した場合の対処法について適切に訓練されていることを確認します。
検証された滅菌プロセスは高い信頼性を提供しますが、継続的な保証には堅牢な品質管理プログラムが不可欠です。これには、最終製品のテストと、それが製造される環境の監視が含まれます。
無菌試験は、滅菌製品のバッチ中の生存微生物の存在を検出するための重要な最終品質チェックです。 USP<71> などの規制に従って、主に 2 つの方法があります。
膜濾過: これは濾過可能な製品に推奨される方法です。液体製品は、孔径 0.45 ミクロン以下の滅菌膜フィルターを通過します。これにより、フィルター表面の微生物が捕捉されます。次に、膜をすすぎ、微生物の増殖を阻害する可能性のある生成物残留物を除去し、2 つの半分に切断し、2 つの異なる種類の培地 (大豆カゼイン消化培地と流動チオグリコール酸培地など) に入れて、広範囲の好気性微生物、嫌気性微生物、および真菌微生物を検出します。
直接接種: この方法は、オイル、懸濁液、医療機器など、濾過できない製品に使用されます。製品の一部を 2 種類の異なる培地に直接添加します。
どちらの方法も 14 日間のインキュベーション期間を必要とし、その間に培地の微生物の増殖 (濁り) を目視検査します。増殖がないことは、結果が無菌であることを示します。
環境モニタリング (EM) は、製造環境の微生物および粒子の清浄度を評価する予防的な品質管理手段です。これは、無菌調製における GMP 準拠の重要な部分です。
空気モニタリング:これは、寒天プレート上で既知の量の空気を吸引するアクティブエアサンプラーを使用するか、一定期間空気にさらされた沈降プレートを使用することによって行われます。次に、これらを培養して、単位空気あたりの生存可能な微生物の数を測定します。
表面モニタリング: 綿棒またはコンタクト プレート (表面に押し付けられた寒天プレート) は、作業面、機器、壁などのクリーンルーム内の表面をサンプリングするために使用されます。
従業員のモニタリング: 従業員が汚染源になっていないことを確認するために、手袋、ガウン、その他の人員の表面をサンプリングします。
環境モニタリングからのデータは傾向分析を提供し、潜在的なリスクを特定し、製品が影響を受ける前に調査を開始するのに役立ちます。
最先端の滅菌器やクリーンルームであっても、訓練を受けた担当者がなければ役に立ちません。無菌技術は、微生物の侵入を防ぐために制御された環境で実行される一連の手法です。これは、無菌製剤、特に最終滅菌ではなく無菌処理される製品の重要な成分です。
無菌調製に関わるすべての職員に対する適切なトレーニングは交渉の余地のないものです。このトレーニングは包括的なものであり、次の内容が含まれる必要があります。
適切な手指衛生とガウンの手順。
滅菌材料および器具の正しい取り扱い。
無菌フィールドを維持し、相互汚染を防ぎます。
クリーンルームの目的と空気の流れのダイナミクスを理解する。
定期的な再認定とメディア充填テストにより、能力を確認します。
このトレーニングは 1 回限りのイベントではありません。継続的な教育と品質の文化が汚染に対する最善の防御策です。
無菌試験方法の比較
| 方法 | 適用対象 | 利点 | 制限事項 |
| 膜ろ過 | 濾過可能な液体、大量 | 高 sensitivity; tests the entire sample; removes product inhibitors. | より複雑で労働集約的。特殊な機器が必要です。膜が詰まる危険性があります。 |
| 直接接種 | 少量の濾過不可能な製品 | シンプルかつ直接的。基本的な実験器具が必要です。セットアップにかかる時間が短縮されます。 | サンプル量が限られているため感度が低い。製品は微生物の増殖を阻害する可能性があります。 |
無菌調製に携わる組織にとって、厳格な規制要件を遵守することは交渉の余地がありません。これらの規格は最終製品の安全性を保証し、堅牢な無菌保証プログラムの基礎となります。
適正製造基準 (GMP) は、製品が品質基準に従って一貫して生産および管理されていることを保証する一連の規制とガイドラインです。滅菌に関しては、米国の 21 CFR Part 211 に記載されているような GMP 規制により、すべてのプロセスが検証され、適切に管理される必要があると規定されています。滅菌に関する主な GMP 要件には以下が含まれます。
プロセスの検証: 滅菌プロセスは、必要な無菌保証レベル (SAL) を一貫して達成することが証明されなければなりません。これには、設置適格性評価 (IQ)、運用適格性評価 (OQ)、および性能適格性評価 (PQ) のための書面による計画とレポートが含まれます。
機器と設備: 滅菌機器は適切に設計、保守、および校正されている必要があります。施設およびクリーンルームの環境は、滅菌前に生物負荷を最小限に抑えるために制御する必要があります。
文書化: 日付、時刻、サイクルパラメータ、オペレーター名など、各滅菌サイクルの詳細な記録を維持し、バッチごとに確認する必要があります。
最終滅菌の優先: GMP ガイダンスでは、無菌処理よりも高いレベルの無菌保証が提供されるため、可能な限り最終滅菌 (最終容器での滅菌) を強く推奨しています。
米国薬局方 (USP) には、無菌製剤と無菌保証に関する詳細なガイダンスを提供する一連の章があり、FDA などの規制機関によってよく引用されています。
USP <71> 無菌試験: この章では、医薬品、生物製剤、医療機器の無菌性を試験するための公定法の概要を説明します。膜濾過と直接接種法の両方の手順について詳しく説明します。
USP <797> 医薬品の配合 - 無菌製剤: この章では、薬局における無菌薬剤の配合に関する具体的なガイドラインを提供します。施設の設計や人材トレーニングから、品質保証、使用後の日付まで、あらゆるものをカバーしています。
USP <1211> 公定記事の滅菌および滅菌保証: この情報章では、方法、検証、滅菌保証の概念など、滅菌の原則の包括的な概要を説明します。これは、無菌性が絶対的なものではなく、SAL として表される確率であることを裏付けています。
国際標準化機構 (ISO) は、ヘルスケア製品の滅菌に関する国際的に認められた基準を発行しています。これらの基準に準拠することで、多くの場合、さまざまな国の規制当局の承認が合理化されます。
ISO11135: この規格は、医療機器のエチレンオキシド滅菌プロセスの開発、検証、日常管理の要件を指定します。
ISO 11137: この一連の規格(パート 1 ~ 3)はヘルスケア製品の放射線滅菌を対象としており、ガンマ線、電子ビーム、X 線照射などの方法を使用したプロセス開発、検証、日常管理の要件を詳述しています。
ISO17665: この規格は、ヘルスケア製品の湿熱滅菌プロセスの開発、検証、日常管理のためのフレームワークを提供します。オートクレーブを検証するための基礎としてよく使用されます。
ISO 13408: この一連の規格は無菌処理に焦点を当てており、最終滅菌できない製品にとって重要です。製造全体を通じて無菌性を維持するためのプロセス、設備、機器の設計に関するガイダンスを提供します。
滅菌基準の比較
| 規格・規制 | 重点領域 | 主な要件 | 主な用途 |
| GMP (21 CFR パート 211) | 製造全般の管理 | すべてのプロセス (IQ/OQ/PQ) の検証、適切な文書化、終末滅菌の優先事項。 | 医薬品および医療機器の製造。 |
| USP <71> | 完成品のテスト | 無菌試験の特定の方法 (膜濾過、直接接種) では、14 日間のインキュベーションが必要です。 | 無菌製品の品質管理。 |
| ISO11135 | エチレンオキサイド滅菌 | 詳細なプロセス検証、ガス濃度、温度、湿度、暴露時間の制御。 | 熱/湿気に敏感な医療機器。 |
| ISO 17665 | 湿熱殺菌 | 物理的および生物学的パラメータを含む、蒸気滅菌サイクルの開発および検証のためのガイドライン。 | 汎用オートクレーブ対応、耐熱性のある製品です。 |
無菌調製の分野は、効率の向上、安全性の向上、環境への影響の削減の必要性によって常に進化しています。技術の進歩により、より洗練された持続可能なソリューションが生まれています。
従来の方法を超えて、特に機密性の高い素材に関しては、新しい技術が注目を集めています。
蒸発過酸化水素 (VHP): VHP 滅菌は、その低温操作、短いサイクル時間、プラスチックや電子機器を含む幅広い材料との適合性により、ますます人気が高まっています。アイソレータ、クリーンルーム、医療機器の滅菌によく使用されます。
放射線滅菌: ガンマ線と電子線 (E ビーム) を含むこの方法は、非常に効果的な「低温」滅菌プロセスです。使い捨て医療機器や医薬品の大量滅菌に最適です。製品を包装した後に滅菌することで、滅菌後の汚染のリスクを最小限に抑えることができるという明らかな利点があります。
オゾン滅菌: オゾンは滅菌剤として使用できる強力な酸化剤です。現場で生成されるため、有毒化学物質の保管や取り扱いの必要がなく、分解されて酸素に戻るため、環境に優しい選択肢となります。
自動化により、前洗浄から最終包装に至る滅菌ワークフローが変革されています。
ロボットハンドリングシステム: 器具や材料の取り扱いと輸送にロボットが導入されており、人為的ミスや汚染のリスクが軽減されています。これは滅菌処理部門で特に価値があり、滅菌器の積み下ろしや器具の仕分けを自動化できます。
自動再処理: 統合システムは、洗浄、消毒、滅菌機能を単一の自動化されたワークフローに統合します。これにより、プロセスが合理化され、スループットが向上し、すべてのサイクルで一貫した検証済みのプロセスが保証されます。
データ分析とIoT: 最新の滅菌器には、サイクルパラメータをリアルタイムで監視できるセンサーとソフトウェアが装備されています。このデータは、パフォーマンスの追跡、メンテナンスの必要性の予測、完全なトレーサビリティとコンプライアンスのためのデジタル記録の提供に使用できます。
持続可能性に世界的に焦点を当てている滅菌業界は、環境フットプリントを削減する方法を模索しています。
エネルギーと水の消費量の削減: メーカーは、より効率的な加熱および給水システムを備えた滅菌器を設計しています。オートクレーブ用の水再循環システムなどの革新により、サイクルあたりの水の使用量を大幅に削減できます。
代替滅菌剤: 業界は、エチレンオキシド (EtO) など、既知の環境リスクや健康リスクを伴う滅菌剤から遠ざけようとしています。 VHP のようなより新しく持続可能な代替品は、有毒な残留物を残さず、無害な副産物に分解されます。
持続可能な包装: 滅菌製品には生分解性でリサイクル可能な包装材料の使用が増加しています。これにより、使い捨てデバイスやパッケージに関連する廃棄物の削減に役立ちます。
これらの傾向は、無菌製剤の未来を形作る、よりスマートで安全、より環境に配慮した滅菌方法への移行を浮き彫りにしています。

滅菌器は、脈動真空、冷気干渉除去、真空乾燥機能を備えた飽和純粋蒸気を滅菌媒体として湿熱滅菌を行います。 滅菌衣類、器具、工具、ゴム栓、アルミキャップ、飼料、廃棄物、非腐食性液体の滅菌に幅広...
詳細を見る
滅菌器は、脈動真空、冷気干渉除去、真空乾燥機能を備えた飽和純粋蒸気を滅菌媒体として湿熱滅菌を行います。 特殊な密閉隔離構造を追加し、装置の前端と後端の厳密な生物学的隔離と密閉を実現し、3段階の...
詳細を見る
低温(60℃または78℃)の蒸気とホルムアルデヒドの混合ガスを真空条件下で急速に浸透させることにより、通気性のある滅菌包装器具(内腔や隙間などの複雑な構造を含む)を迅速かつ効率的に滅菌することが...
詳細を見るサニタリーグレード滅菌器の紹介: 現代の滅菌における定義と重要性 サニタリーグレード滅菌器の定義と中心概念: 衛生グレードの滅菌器 は、重要な産業環境で必要とされる最高レベルの微生物制御を達成および維持するように設計された、高度に特殊化された滅菌装置です。一般的な滅菌器とは異なり、衛生グレードの滅菌器は、微生物......
続きを読むはじめに 今日のペースの速い世界では、ヘルスケア、製薬、食品産業で使用される製品と材料の安全性と無菌性を確保することが最も重要です。汚染は悲惨な結果をもたらし、深刻な健康リスク、高額なリコール、さらには生命を脅かす状況につながる可能性があります。その結果、滅菌プロセスは堅牢で信頼性が高く、一貫して効果的でなければなりません。さまざまな分野で広く採用されてい......
続きを読むの紹介 滅菌準備滅菌器 無菌製剤とは何ですか? 無菌調製とは、生存微生物を含まない製品である無菌製品を配合または製造するプロセスを指します。これは、注射剤、点滴静注液、点眼液など、人体への投与を目的とした製品を製造する業界では重要なプロセスです。また、手術やその他の処置で使用される滅菌医療機器や器具を準備す......
続きを読むなぜすべての医薬品に滅菌製剤用の滅菌装置が必要なのでしょうか? 現代の医学と医薬品では、無菌処理が非常に重要です。研究室での研究、医薬品の製造、あるいは病院での臨床使用のいずれにおいても、すべての医薬品とすべての医療機器は、その安全性と有効性を確保するために厳格な無菌処理を受けなければなりません。医薬品や医療機器を滅菌するための重要な機器として、 ......
続きを読む



